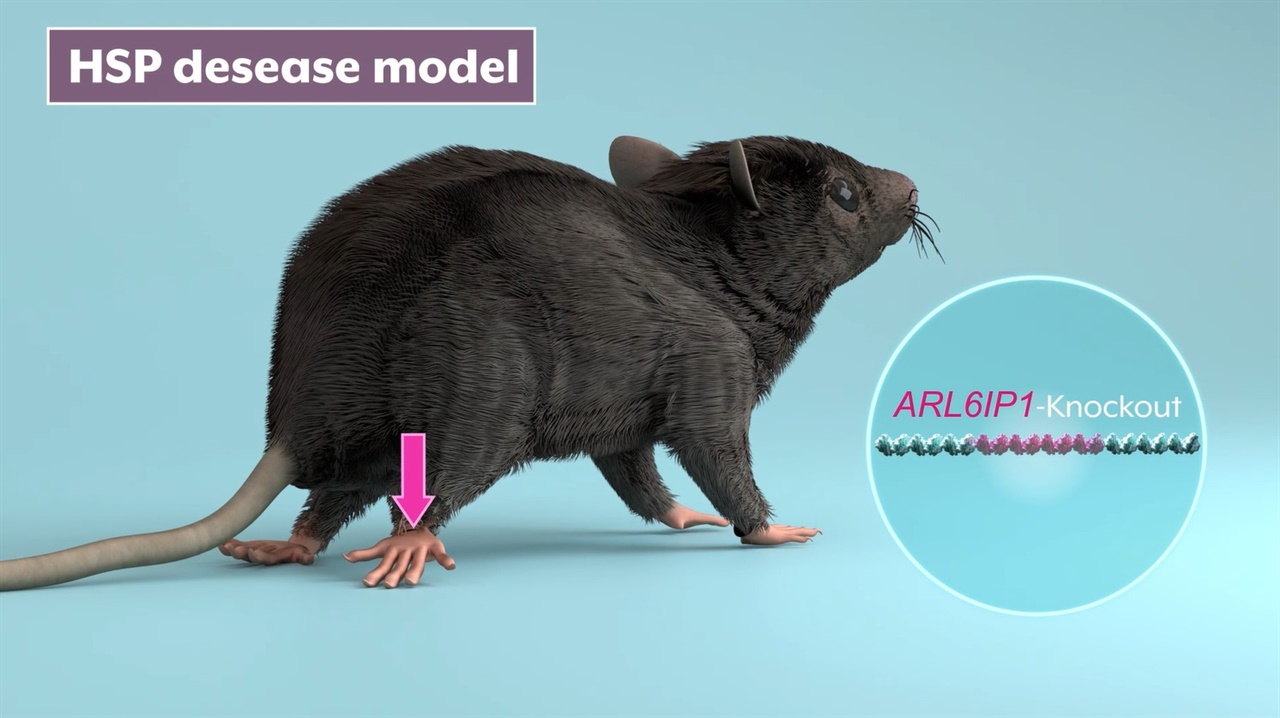
▲'유전성 하지강직성 대마비 증후군(HSP)' 질환을 앓는 실험쥐 모델. ⓒ 한국생명공학연구원 제공
희귀난치성 유전성 신경계 질환으로 다리 근육이 점차 약해져 마비되고, 근육의 긴장성이 증가하며, 뻣뻣해지져 마비에 이르게 되는 '유전성 하지강직성 대마비 증후군(Hereditary Spastic paraplegia, 아래 HSP)'에 대한 유전자치료 기술을 국내 연구진이 개발하는데 성공했다.
이로써 아직까지 근본적 치료제가 없던 HSP 환자들에게 새로운 희망이 될 수 있을 것으로 기대된다.
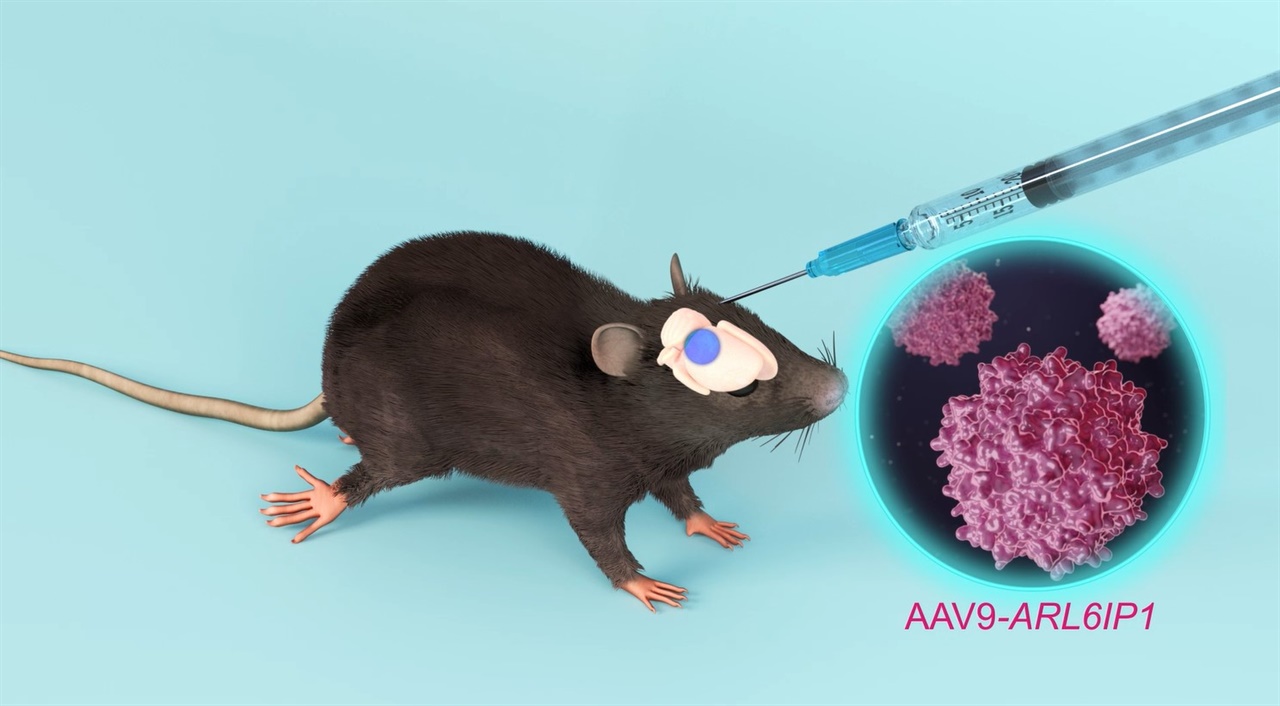
▲'유전성 하지강직성 대마비 증후군(HSP)' 질환을 앓는 실험쥐 모델에 ARL6IP1을 아데노 부속 바이러스(Adeno Associated Virus, AAV) 전달체에 탑재해 만든 유전자 치료제를 투여하고 있다. ⓒ 한국생명공학연구원 제공
한국생명공학연구원(원장 김장성, 아래 생명연)은 24일 "줄기세포연구센터 정초록 박사 연구팀이 HSP에 대한 유전자치료 기술을 개발하는데 성공했다"면서 "연구팀은 이러한 기전을 바탕으로 HSP에 대한 유전자치료 기술을 개발하고, 최초로 동물모델에서 효능검증까지 마쳤다"고 밝혔다.
HSP는 전 세계적으로 10만 명당 1.8명꼴로 발생하는 희귀난치성 질환으로 중추신경계 신경퇴행성 질환에 해당한다. 발병 원인으로 80여 종의 유전자가 복잡하게 관여하는 것으로 알려져 치료제 개발이 어려우며 대표적 증상인 하지 강직성과 근 손실에 대한 증상 완화에 치중하는 실정이라고 한다.
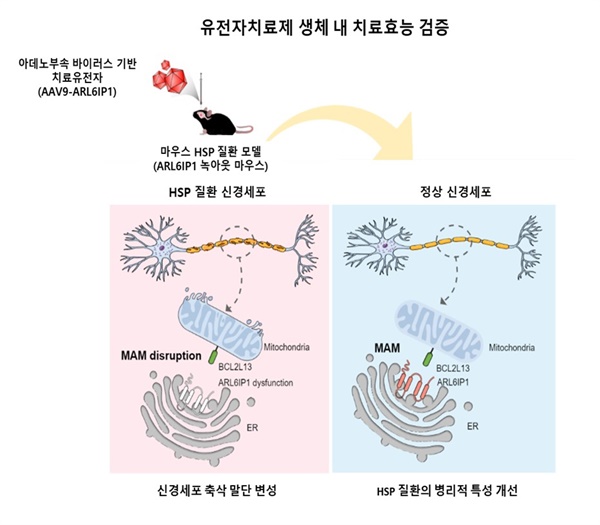
▲유전성 하지강직성 대마비 증후군의 원인 유전자인 ARL6IP1 유전자의 변이에 따른 병인 기전 규명 및 아데노부속 바이러스 기반 유전자 치료제 치료효능 검증 모식도 ⓒ 한국생명공학연구원 제공
이에 연구팀은 특정 유전자인 'ARL6IP1(ADP ribosylation factor like GTPase 6 interacting protein 1)' 유전자에 의해 HSP가 발병한다는 메커니즘을 이번에 새롭게 밝혀냈다.
우선, 마우스 질환 모델에서 ARL6IP1이 미토콘드리아 연결 소포체 막(Mitochondria-Associated ER Membrane, MAM)에 존재하면서 세포소기관의 항상성에 관여하여 신경염증에 의한 신경세포 손상을 조절한다는 사실을 확인했다. 미토콘드리아 연관 소포체 막(MAM)은 미토콘드리아와 소포체가 물리적으로 맞닿아 있는 부분으로, 자가포식, 지질합성, 칼슘수송 등의 세포 내 신호전달이 오가는 허브라고 한다.
즉, ARL6IP1의 기능상실이 유발한 '자가포식' 조절 이상으로 인해 손상된 미토콘드리아가 신경세포에 축적되면 신경 퇴행이 발생해 HSP가 발병한다는 것이다.
참고로 자가포식 작용(Autophagy)이란, 세포 내에서 대사와 합성을 통해 만들어진 물질이 쓰임을 다하거나 산화 스트레스와 같은 요인으로 인해 내부 기관이 손상을 받았을 때 라이소좀에서 이를 분해하는데 이 과정을 '자가포식'이라고 한다.
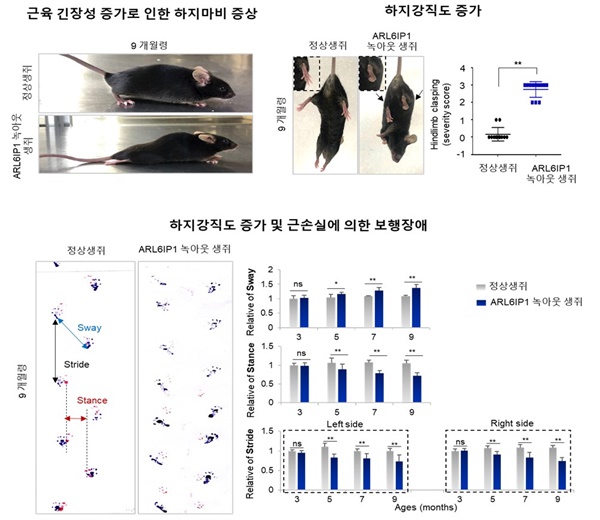
▲HSP 환자 질환표현형을 모방하는 마우스 질환 동물모델 구축 ⓒ 한국생명공학연구원 제공
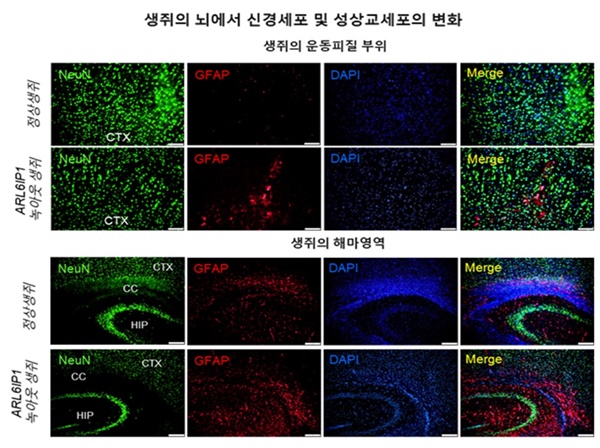
▲ARL6IP1 유전자를 제거한 마우스에서 HSP 환자의 대표적 질환 표현형인 다리 근육이 점차 약해져 마비되고, 근육의 긴장성과 하지 강직도의 증가를 확인함. 또한, 활성화된 성상교세포의 증가가 두드러지게 나타남 ⓒ 한국생명공학연구원 제공
나아가 연구팀은 이를 바탕으로 HSP에 대한 유전자치료 기술을 개발하고, 최초로 실험쥐 모델을 통해 효능을 검증해냈다.
ARL6IP1을 아데노 부속 바이러스(Adeno Associated Virus, AAV) 전달체에 탑재해 유전자 치료제를 만들고, 이렇게 만든 유전자 치료물질을 실험쥐에 투여했다. 그 결과, 치료를 받은 HSP 질환 마우스는 하지 강직성이 감소하고, 보행장애가 호전되었을 뿐만 아니라 뇌 조직상 병변과 신경염증 반응도 개선됐다.
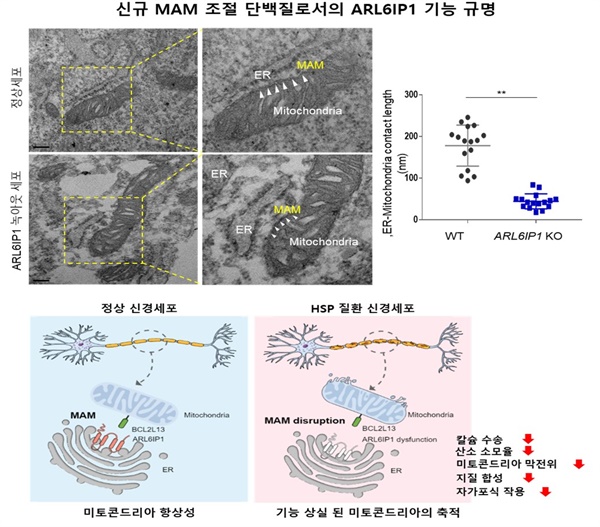
▲HSP 질환의 원인유전자 ARL6IP1의 MAM 조절 단백질로서의 신규 기능 규명ARL6IP1 유전자가 미토콘드리아 연관 소포체 막(Mitochondria-Associated ER Membrane, MAM) 특정 부위에 위치하는 신규 MAM 단백질임을 규명하고 신경세포 내 지질합성, 자가포식 등의 핵심기능을 조절함을 확인하였음. 이러한 ARL6IP1 기능상실은 MAM의 기능 이상을 초래하여 신경 퇴행을 유발하는 병인 기전을 규명함. ⓒ 한국생명공학연구원 제공
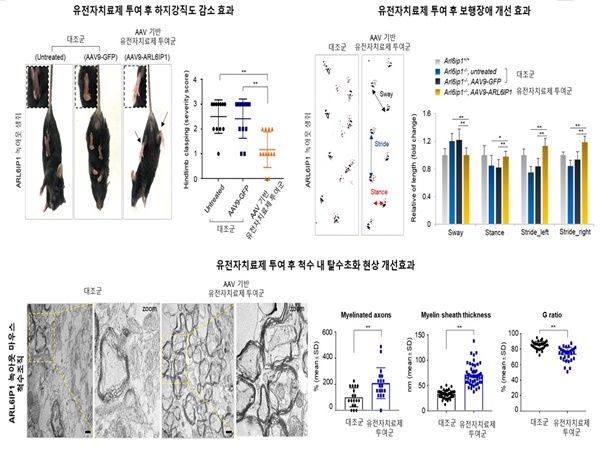
▲아데노부속 바이러스(AAV) 기반 유전자 치료제 치료효능 검증AAV 기반 유전자 치료제(AAV9-ARL6IP1)를 마우스 HSP 질환모델의 뇌 일차성 운동 피질 영역에 미세주입한 결과, HSP 질환 표현형인 하지 강직도 감소와 보행장애 개선, 척수 조직 내 탈수초화 현상 개선 효과를 확인함. ⓒ 한국생명공학연구원 제공
연구책임자인 정초록 박사는 "HSP에 대한 새로운 기전을 제시하고 유전자치료 가능성을 열었다는데 큰 의미가 있다"면서 "이번 연구성과에 큰 도움이 된 생명연의 '타겟 도출→기전검증→효능검증' 유전자 치료제 개발 파이프라인이 국내 희귀질환 유전자 치료제 개발에 널리 활용되길 기대한다"고 말했다.

▲HSP를 앓는 쥐에 유전자치료제를 투여하자 보행능력이 개선됐다. ⓒ 한국생명공학연구원 제공
그동안 유전체 기술의 발전으로, 치료방법이 없던 유전성 희귀 난치질환에서도 첨단바이오 의약품이 속속 개발되고 있어 환자와 시장의 큰 주목을 받고 있는 상황이다. '유전자 치료제(gene therapy)'는 환자의 비정상 유전자를 정상 유전자로 바꿔 유전적 결함을 치료하는 의약품으로, 2012년 유전자 결핍에 의한 가족성 고지혈증 치료제인 글리베라(Glybera)가 처음 승인된 이후로 유전자 전달체에 대한 안전성, 효율성 개선 연구를 통해 다양한 희귀난치성 질환을 대상으로 사용 가능성이 확장되고 있다고 연구팀이 부연했다.
한편, 이번 연구는 과학기술정보통신부(장관 이종호) 다부처유전체기술개발사업, 개인기초연구사업, 바이오‧의료기술개발사업 및 생명연 주요사업의 지원으로 수행됐다. 연구 결과는 1월 1일 발간된 의학 분야의 유수 저널인 <Journal of Experimental Medicine(IF 15.3)> 최신 호에 게재됐다.

▲연구진 사진. 뒷줄 왼쪽부터 두 번째 제1저자 임정화 박사, 세 번째 연구책임자 정초록 박사. ⓒ 한국생명공학연구원 제공